https://butterlab.imb-mainz.de/bombyxviewer/
Published in BMC Genomics
https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-07088-7 
Background: The process of identifying all coding regions in a genome is crucial for any study at the level of molecular biology, ranging from single-gene cloning to genome-wide measurements using RNA-seq or mass spectrometry. While satisfactory annotation has been made feasible for well-studied model organisms through great efforts of big consortia, for most systems this kind of data is either absent or not adequately precise.
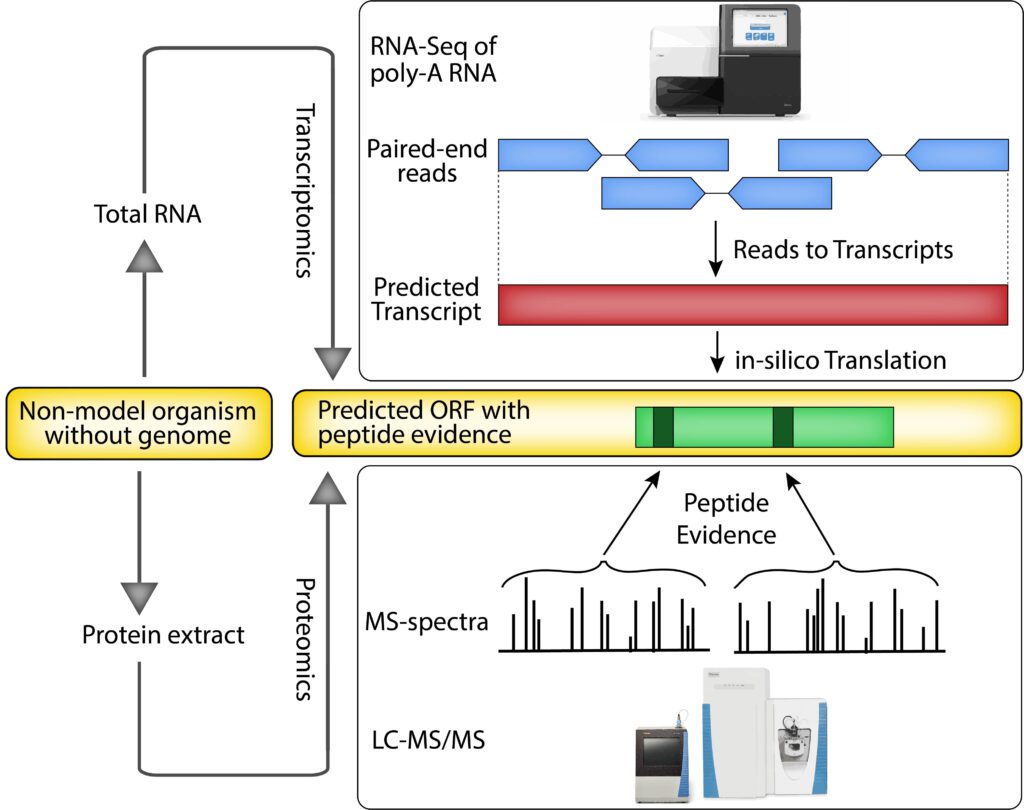
Results: Combining in-depth transcriptome sequencing and high resolution mass spectrometry, we here use proteotranscriptomics to improve gene annotation of protein-coding genes in the Bombyx mori cell line BmN4 which is an increasingly used tool for the analysis of piRNA biogenesis and function. Using this approach we provide the exact coding sequence and evidence for more than 6200 genes on the protein level. Furthermore using spatial proteomics, we establish the subcellular localization of thousands of these proteins. We show that our approach outperforms current Bombyx mori annotation attempts in terms of accuracy and coverage.
Conclusions: We show that proteotranscriptomics is an efficient, cost-effective and accurate approach to improve previous annotations or generate new gene models. As this technique is based on de-novo transcriptome assembly, it provides the possibility to study any species also in the absence of genome sequence information for which proteogenomics would be impossible.